Python 官方文档:入门教程 => 点击学习
引言 在基因组分析中,我们经常会有这么一个需求,就是在一个fasta文件中提取一些序列出来。有时这些序列是一段完整的序列,而有时仅仅为原fasta文件中某段序列的一部分。特别是当数据
在基因组分析中,我们经常会有这么一个需求,就是在一个fasta文件中提取一些序列出来。有时这些序列是一段完整的序列,而有时仅仅为原fasta文件中某段序列的一部分。特别是当数据量很多时,使用肉眼去挑选序列会很吃力,那么这时我们就可以通过简单的编程去实现了。
例如此处在网盘附件中给定了某物种的全基因组序列(0-refer/ Bacillus_subtilis.str168.fasta),及其基因组gff注释文件(0-refer/ Bacillus_subtilis.str168.gff)。
假设在这里我们对该物种进行研究,通过gff注释文件中的基因功能描述字段,加上对相关资料的查阅等,定位到了一些特定的基因。
接下来我们期望基于gff文件中对这些基因位置的描述,在全基因组序列fasta文件中将这些基因找到并提取出来,得到一个新的fasta文件,新文件中只包含目的基因序列。
请使用python3编写一个可以实现该功能的脚本。
一个示例脚本如下(可参见网盘附件“seq_select1.py”)。
为了实现以上目的,我们首先需要准备一个txt文件(以下称其为list文件,示例list.txt可参见网盘附件),基于gff文件中所记录的基因位置信息,填入类似以下的内容(列与列之间以tab分隔)。
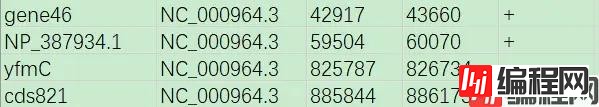
#下列内容保存到list.txt
gene46 NC_000964.3 42917 43660 +
NP_387934.1 NC_000964.3 59504 60070 +
yfmC NC_000964.3 825787 826734 -
cds821 NC_000964.3 885844 886173 -
第1列,给所要获取的新序列命个名称;
第2列,所要获取的序列所在原序列ID;
第3列,所要获取的序列在原序列中的起始位置;
第4列,所要获取的序列在原序列中的终止位置;
第5列,所要获取的序列位于原序列的正链(+)或负链(-)。
之后根据输入文件,即输入fasta文件及记录所要获取序列位置的list文件中的内容,编辑py脚本。
打开fasta文件“Bacillus_subtilis.scaffolds.fasta”,使用循环逐行读取其中的序列id及碱基序列,并将每条序列的所有碱基合并为一个字符串;将序列id及该序列合并后的碱基序列以字典的形式存储(字典样式{'id':'base'})。
打开list文件“list.txt”,读取其中的内容,存储到字典中。字典的键为list文件中的第1列内容;字典的值为list文件中第2-5列的内容,并按tab分割得到一个列表,包含4个字符分别代表list文件中第2-5列的信息)。
最后根据读取的list文件中序列位置信息,在读取的基因组中截取目的基因序列。由于某些基因序列可能位于基因组负链中,需取其反向互补序列,故首先定义一个函数rev(),用于在后续调用得到反向互补序列。在输出序列名称时,还可选是否将该序列的位置信息一并输出(name_detail = True/False)。
<pre class="r" style="overflow-wrap: break-Word; font-style: nORMal; font-variant-ligatures: normal; font-variant-caps: normal; font-weight: 400; letter-spacing: normal; orphans: 2; text-align: start; text-indent: 0px; text-transform: none; widows: 2; word-spacing: 0px; -WEBkit-text-stroke-width: 0px; text-decoration-style: initial; text-decoration-color: initial; margin-top: 0px; margin-bottom: 10px; padding: 9.5px; border-radius: 4px; background-color: rgb(245, 245, 245); box-sizing: border-box; overflow: auto; font-size: 13px; line-height: 1.42857; color: rgb(51, 51, 51); word-break: break-all; border: 1px solid rgb(204, 204, 204); font-family: "Times New Roman";">#!/usr/bin/env python3
# -*- coding: utf-8 -*-
#初始传递命令
input_file = 'Bacillus_subtilis.str168.fasta'
list_file = 'list.txt'
output_file = 'gene.fasta'
name_detail = True
##读取文件
#读取基因组序列
seq_file = {}
with open(input_file, 'r') as input_fasta:
for line in input_fasta:
line = line.strip()
if line[0] == '>':
seq_id = line.split()[0]
seq_file[seq_id] = ''
else:
seq_file[seq_id] += line
input_fasta.close()
#读取列表文件
list_dict = {}
with open(list_file, 'r') as list_table:
for line in list_table:
if line.strip():
line = line.strip().split('\t')
list_dict[line[0]] = [line[1], int(line[2]) - 1, int(line[3]), line[4]]
list_table.close()
##截取序列并输出
#定义函数,用于截取反向互补
def rev(seq):
base_trans = {'A':'T', 'C':'G', 'T':'A', 'G':'C', 'N':'N', 'a':'t', 'c':'g', 't':'a', 'g':'c', 'n':'n'}
rev_seq = list(reversed(seq))
rev_seq_list = [base_trans[k] for k in rev_seq]
rev_seq = ''.join(rev_seq_list)
return(rev_seq)
#截取序列并输出
output_fasta = open(output_file, 'w')
for key,value in list_dict.items():
if name_detail:
print('>' + key, '[' + value[0], value[1] + 1, value[2], value[3] + ']', file = output_fasta)
else:
print('>' + key, file = output_fasta)
seq = seq_file['>' + value[0]][value[1]:value[2]]
if value[3] == '+':
print(seq, file = output_fasta)
elif value[3] == '-':
seq = rev(seq)
print(seq, file = output_fasta)
output_fasta.close()</pre>
编辑该脚本后运行,输出新的fasta文件“gene.fasta”,其中的序列即为我们所想要得到的目的基因序列。
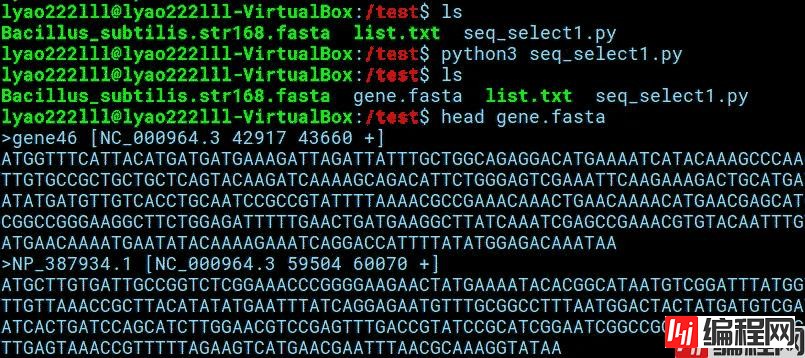
网盘附件“seq_select.py”为添加了命令传递行的Python3脚本,可在shell中直接进行目标文件的I/O处理。该脚本可指定输入fasta序列文件以及记录有所需提取序列位置的列表文件,输出的新fasta文件即为提取出的序列。
#!/usr/bin/env python3
# -*- coding: utf-8 -*-
#导入模块,初始传递命令、变量等
import argparse
parser = argparse.ArgumentParser(description = '\n该脚本用于在基因组特定位置截取序列,需额外输入记录有截取序列信息的列表文件', add_help = False, usage = '\npython3 seq_select.py -i [input.fasta] -o [output.fasta] -l [list]\npython3 seq_select.py --input [input.fasta] --output [output.fasta] --list [list]')
required = parser.add_argument_group('必选项')
optional = parser.add_argument_group('可选项')
required.add_argument('-i', '--input', metavar = '[input.fasta]', help = '输入文件,fasta 格式', required = True)
required.add_argument('-o', '--output', metavar = '[output.fasta]', help = '输出文件,fasta 格式', required = True)
required.add_argument('-l', '--list', metavar = '[list]', help = '记录“新序列名称/序列所在原序列ID/序列起始位置/序列终止位置/正链(+)或负链(-)”的文件,以 tab 作为分隔', required = True)
optional.add_argument('--detail', action = 'store_true', help = '若该参数存在,则在输出 fasta 的每条序列 id 中展示序列在原 fasta 中的位置信息', required = False)
optional.add_argument('-h', '--help', action = 'help', help = '帮助信息')
args = parser.parse_args()
##读取文件
#读取基因组序列
seq_file = {}
with open(args.input, 'r') as input_fasta:
for line in input_fasta:
line = line.strip()
if line[0] == '>':
seq_id = line.split()[0]
seq_file[seq_id] = ''
else:
seq_file[seq_id] += line
input_fasta.close()
#读取列表文件
list_dict = {}
with open(args.list, 'r') as list_file:
for line in list_file:
if line.strip():
line = line.strip().split('\t')
list_dict[line[0]] = [line[1], int(line[2]) - 1, int(line[3]), line[4]]
list_file.close()
##截取序列并输出
#定义函数,用于截取反向互补
def rev(seq):
base_trans = {'A':'T', 'C':'G', 'T':'A', 'G':'C', 'a':'t', 'c':'g', 't':'a', 'g':'c'}
rev_seq = list(reversed(seq))
rev_seq_list = [base_trans[k] for k in rev_seq]
rev_seq = ''.join(rev_seq_list)
return(rev_seq)
#截取序列并输出
output_fasta = open(args.output, 'w')
for key,value in list_dict.items():
if args.detail:
print('>' + key, '[' + value[0], value[1] + 1, value[2], value[3] + ']', file = output_fasta)
else:
print('>' + key, file = output_fasta)
seq = seq_file['>' + value[0]][value[1]:value[2]]
if value[3] == '+':
print(seq, file = output_fasta)
elif value[3] == '-':
seq = rev(seq)
print(seq, file = output_fasta)
output_fasta.close()
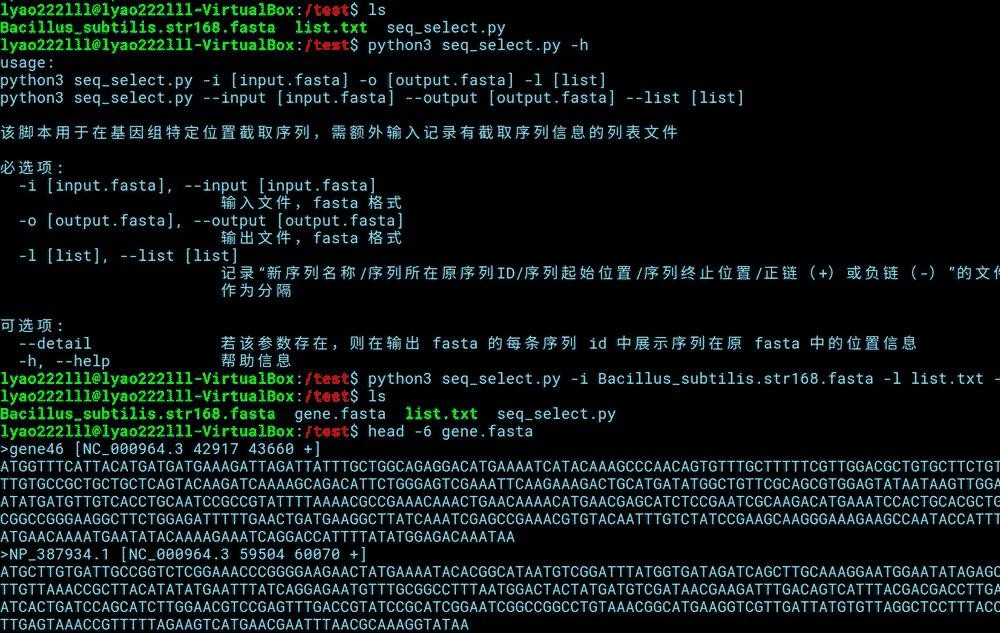
适用上述示例中的测试文件,运行该脚本的方式如下。
#python3 seq_select.py -h
python3 seq_select.py -i Bacillus_subtilis.str168.fasta -l list.txt -o gene.fasta --detail
源码提取链接: https://pan.baidu.com/s/1kUhBTmpDonCskwmpNIJPkA?pwd=ih9n
提取码: ih9n
以上就是使用Python脚本提取基因组指定位置序列的详细内容,更多关于python提取基因组位置序列的资料请关注编程网其它相关文章!
--结束END--
本文标题: 使用Python脚本提取基因组指定位置序列
本文链接: https://lsjlt.com/news/119043.html(转载时请注明来源链接)
有问题或投稿请发送至: 邮箱/279061341@qq.com QQ/279061341
2024-03-01
2024-03-01
2024-03-01
2024-02-29
2024-02-29
2024-02-29
2024-02-29
2024-02-29
2024-02-29
2024-02-29
回答
回答
回答
回答
回答
回答
回答
回答
回答
回答


官方手机版

微信公众号

商务合作

0